
 This issue in
This issue in 
Exploring Genomes with the Self-Organizing Map
by Shaun Mahony and Aaron Golden
The use of pattern recognition software is not new in the field of bioinformatics, but is perhaps not as developed as it should be. With the growing amounts of data that are being produced by various micro-array technologies and other devices on the one hand, and an appreciation of the fact that there is more to the vast amount of 'non-coding' DNA than meets the eye certainly for H. sapiens on the other, an ability to apply unsupervised algorithms to winnow down the enormous variety of parameter space to a sub-domain that is contextually meaningful is becoming more and more relevant. At the National Centre for Biomedical Engineering Science, National University of Ireland, Galway researchers have been exploring the capabilities of the Self-Organizing Map (SOM) algorithm to this end.
The Self-Organizing Map (SOM) neural network is based around the concept of a lattice of interconnected nodes, each of which contains a model. The models begin as random values, but during the iterative training process they are modified to represent different subsets of the training set. The algorithm effectively performs a map from the high dimensional input vector space to a low dimensional representation whose nodes are characterised by these subsets. Optimum use of the SOM necessitates defining the descriptor that encapsulates the 'pattern' we wish to classify. We have utilised well known descriptors whose representation in the conventional probabilistic frameworks would be difficult if not computationally impracticable - this is one particularly compelling advantage of the SOM algorithm. We highlight two examples of our work to date:
Automatically Generating Multiple Gene Models with RescueNet
The study of codon usage variation in coding as against non-coding regions of DNA has been scrutinised for many years, and whilst there is considerable evidence arguing for clear deviations in usage patterns between the two cases, our actual understanding of the nature of synonymous codon usage is still limited, with the likelihood that more subtle characteristics have yet to be uncovered. Using synonymous codon usage as the descriptor, this SOM variant captures such high dimensional diversity. RescueNet allows the user to identify clusters of genes that have similar codon usage patterns, to identify genes displaying atypical codon usage patterns and perhaps most interestingly, to analyse a contiguous genomic sequence for areas displaying similar codon usage patterns to the major patterns found in a training set, thus making it a valuable annotation tool. In figures 1 and 2 we outline examples of the algorithm's use in these contexts.
|
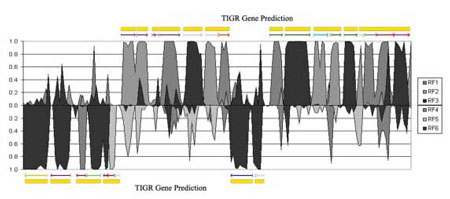
|
Transcription Factor Binding Site Identification with SOMBRERO
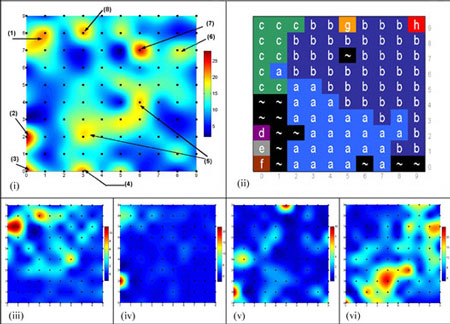
Every eukaryotic genome sequenced thus far has shown vast quantities of DNA which do not appear to contain protein-coding regions. Although such non-coding DNA can play important structural roles, much of it also harbors intricate gene regulatory information, including short (6-20 bp) motifs that serve as transcription factor binding sites (TFBS). Cracking the so-called 'cis-regulatory code' has become an important goal in the decoding of genomic data, and an integral part of this challenge is the identification of TFBS. Their identification however is complicated their short size, the inherent degeneracy, and their location ranging by three orders of magnitude up and downstream of a given gene's transcription start site. On the plus side, there are many incidents of TFBS classes in the genome, and many correspond to fundamental processes common to us and other species, so hunting in conserved regions of DNA between human and say, mouse, would be expected to boost the signal to noise nature of the data so presented. We have developed a SOM algorithm, SOMBRERO, that uses as its descriptor of TFBS diversity the position weight matrix (PWM), perhaps the most effective way to characterise the degenerate set of a given TFBS class. Our initial experiments have indicated that SOMBRERO yields advantageous performance over more conventional probablistic/statistical mechanical techniques. In Figure 3 we show SOMBRERO-Viewer, which allows one to examine the results of the trained SOM.
|
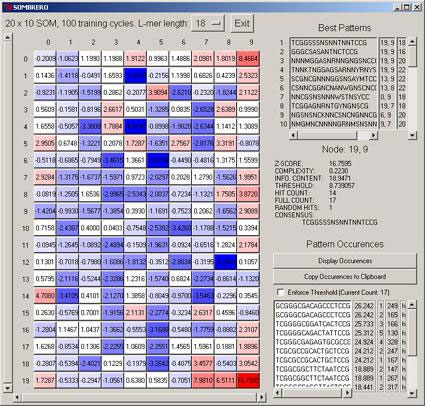
Thus far our work has been devoted to developing working SOM models that are applicable in specific areas and that function on a par with existing probabilistic/statistical algorithms. We aim to go further and to incorporate the SOM algorithms into more contextually defined formalisms, such as the known clustering of TFBS within eukaryotic promoter regions, thus improving the efficacy of the technique. As the data loads facing researchers grow, along with our appreciation of the inherent complexity of regulatory networks we need to decode, we believe appropriate applications of the SOM algorithm will become more and more necessary.
Link:
http://bioinf.nuigalway.ie/shaun.html
Please contact:
Shaun Mahony, National Centre for Biomedical Engineering Science,
National University of Ireland, Galway/IUC
Tel: +353 91 512074
E-mail: shaun.mahony![]() nuigalway.ie
nuigalway.ie