

by Hidde de Jong, Michel Page, Céline Hernandez, Hans Geiselmann and Sébastien Maza
In order to understand the functioning of an organism, the network of interactions between genes, mRNAs, proteins, and other molecules needs to be elucidated. Since 1999, researchers in the bioinformatics group at INRIA Rhône-Alpes have been developing a computer tool for the modeling and simulation of genetic regulatory networks in collaboration with molecular biologists.
The sequencing of the entire genome of prokaryotic and eukaryotic organisms has been completed in the past few years, culminating in the presentation of a working draft of the human genome last June. The analysis of these huge amounts of data involves such tasks as the prediction of folding structures of proteins and the identification of genes and regulatory signals. It is clear, however, that the structural analysis of sequence data needs to be complemented with a functional analysis to elucidate the role of genes in controlling fundamental biological processes.
One of the central problems to be addressed is the analysis of genetic regulatory systems controlling the spatiotemporal expression of genes in an organism. The structure of these regulatory systems can be represented as a network of interactions between genes, proteins, metabolites, and other small molecules. The study of genetic regulatory networks will contribute to our understanding of complex processes like the development of a multicellular organism.
In addition to new experimental tools permitting the expression level to be rapidly measured in a massively parallel way, computer tools for the modeling, visualization, and simulation of genetic regulatory systems will be indispensable. Most systems of interest involve many genes connected through cascades and positive and negative feedback loops, so that an intuitive understanding of their dynamics is hard to obtain. As a consequence of the lack of quantitative information on regulatory interactions, traditional modeling and simulation techniques are usually difficult to apply. To counter this problem, we have developed a method for the qualitative simulation of regulatory systems based on ideas from mathematical biology and artificial intelligence.
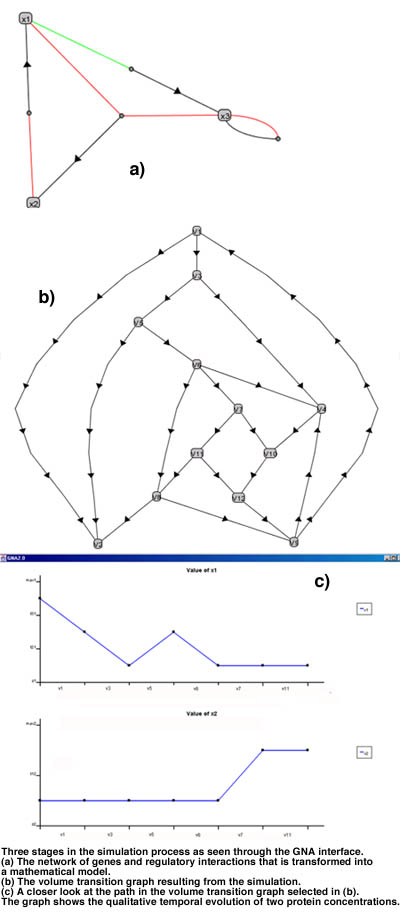
The method describes genetic regulatory systems by piecewise-linear differential equations with favourable mathematical properties. The phase space is subdivided into volumes in which the equations reduce to simple, linear and orthogonal differential equations imposing strong constraints on the local behavior of the system. By analyzing the possible transitions between volumes, an indication of the global behavior of the system can be obtained. In particular, the method determines steady-state volumes and volume cycles that are reachable from an initial volume. The steady-state volumes and volume cycles correspond to functional states of the regulatory system, for instance a response to a physiological perturbation of the organism (a change in temperature or nutrient level).
The above method has been implemented in Java 1.2 in a program called GNA (Genetic Network Analyzer). GNA reads and parses input files with the equations and inequalities specifying the model of the system as well as the initial volume. An inequality reasoner iteratively generates the volumes that are reachable from the initial volume through one or more transitions. The output of the program consists of the graph of all reachable volumes connected by transitions. A graphical interface facilitating the interaction of the user with the program is under development. At present, a visualization module has been realized by which a network of interactions between genes can be displayed, as well as the volume transition graph resulting from the simulation. In addition, the user can focus upon particular paths in the graphs to study the qualitative temporal evolution of gene product concentrations in more detail (see figures).
GNA has been tested using genetic regulatory networks described in the literature, such the example of lambda phage growth control in the bacterium Escherichia coli. Simulation experiments with random regulatory networks have shown that, with the current implementation, our method remains tractable for systems of up to 18 genes involved in complex feedback loops.
We plan GNA to evolve into an environment for the computer-supported analysis of genetic regulatory networks, covering a range of activities in the design and testing of models. These activities, such as the validation of hypothesized models of regulatory networks by means of experimental data, will be accessible through a user-friendly graphical interface. In parallel, we will apply the method to the analysis of bacterial regulatory systems in collaboration with biologists at the Université Joseph Fourier in Grenoble.
Links:
HELIX project: http://www.inrialpes.fr/helix/
Please contact:
Hidde de Jong - INRIA Rhône-Alpes
Tel: +33 4 76 61 53 35
E-mail: Hidde.de-Jong@inrialpes.fr