ERCIM News 64January 2006Contents 
This issue in pdf Subscription Archive: Next issue: April 2006 Next Special theme:
|
by Anna Tonazzini, Francesco Bonchi, Stefania Gnesi, Ercan Kuruoglu and Sergio Bottini
At ISTI-CNR, Pisa, researchers from different areas of computer science are studying an integrated and interdisciplinary approach to various problems in Computational Biology and Bioinformatics.
The achievements of the Human Genome Project and the rapid development of post-genomic technology have dramatically increased the importance of genomics and genetics in disease analysis and diagnosis, novel drug and therapy discovery, and early detection or even prediction of disease. The aim is to improve healthcare strategies and, ultimately, the quality of life of the individual. Due to the enormous flow of heterogeneous biological data that is being made available, powerful tools for storage and retrieval, processing, analysis and modelling are becoming increasingly crucial in order to be able to extract useful knowledge from this data.
Bioinformatics exploits modern computing and communication technology, and is stimulating research that addresses computationally demanding challenges in biology and medicine. This highly interdisciplinary field includes data mining, modelling of complex systems, 2D and 3D visualization, signal and image processing and analysis, 'in silico' modelling and simulation, and algorithms for large-scale combinatorial problems.
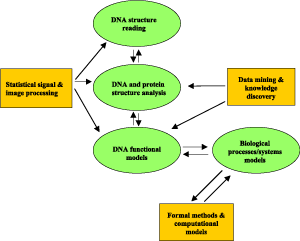
Researchers from the ISTI Laboratories for Signal and Images, Knowledge Discovery and Delivery and Formal Methods and Tools form an interdisciplinary group whose comprehensive research in a number of areas of bioinformatics has been recently formalized in a Work Package of the national CNR project on 'Computational Biology'.
|
Our main goal is the development of models and analysis methods that can help to describe and understand the spatial characteristics of DNA with a functional value, and the computational mechanisms behind complex biological systems such as gene regulatory networks. A bottom-up strategy will be adopted, in which low-level processing integrates with high-level classification and modelling. The focus will be on the structural analysis of genomes and proteins, and on the detection and functional analysis of clusters of genes related to underlying biological processes in microarray experiments.
A large number of genomes, ranging from viral and microbial pathogens to higher organisms, have now been fully sequenced and made publicly available for investigations at various levels. Nevertheless, although DNA sequencing is a mature technique and many research efforts to further improve the algorithmic phase are reported in the literature, accurate identification of bases has not yet been fully achieved by the software of available automatic sequencing machines. In this respect, we are currently studying unsupervised statistical techniques to model electrophoresis signals and correct the colour cross-talk and peak-spreading phenomena. At the genome scale, we have developed efficient algorithms for fragment assembly by partial overlap in the shotgun sequencing method. As per high-level processing, we are working on comparative genomics for the identification of conserved and invariant structural elements with functional value within the genomes. Special attention is being paid to the large portion of non-coding regions.
In proteomics, we take advanced techniques for mining complex and high-dimensional information spaces, and apply them to frequent local pattern discovery in protein databases, and to the alignment of proteins at the various structural levels, with the aim of finding common functional characteristics. Knowledge discovery and representation methods will be then exploited as knowledge-based adaptive systems for decision support in medicine and surgery (eg for studying autoimmunity mechanisms and for compatibility testing in organ transplant surgery).
Thanks to recent advances in microarray technology, we are now able to monitor the activity of a whole genome under multiple experimental conditions. Large amounts of data are becoming available, providing simultaneous measurements of expression profiles and of interactions of thousands of genes. The challenge is to discover the complex functional dependencies underlying these data and to identify biological processes driving gene coregulation. At ISTI, techniques for unsupervised clustering of gene expression maps from microarray data are now being investigated. In particular, we are studying statistical techniques of Blind Source Separation, such as Independent Component Analysis (ICA), nonlinear and constrained ICA, and Dependent Component Analysis, which should provide non-mutually exclusive gene clusters. The results of these analyses will be compared with those of local pattern discovery strategies such as constraint-based mining, and possibly used as input to sophisticated clustering techniques. The ultimate goal is to provide simulations and modelling of molecular interactions and metabolic pathways. In this respect, we are also studying formal methods that can be used to describe complex biological systems and verify their properties. Due to the real and massive parallelism involved in molecular interactions, investigations into the exploitation of biomolecular models as examples of global and parallel computing are also in progress.
The research activity described above is carried out in collaboration with other institutions in the fields of biomedicine and informatics. The biomedical institutions provide us with data and validate the biological significance of the results. Our main collaborations are with the Institute of Biophysics, CNR, Pisa, the National Institute for Applied Sciences (INSA) in Lyon and the Immunohematology Unit, II Pisa Hospital Cisanello. We intend to establish new collaborations with other bioinformatics groups, and in particular we are seeking fruitful interactions within ERCIM.
Link:
http://www.isti.cnr.it/ResearchUnits/Labs/si-lab/ComputationalBiologyGroup.html
Please contact:
Anna Tonazzini, ISTI-CNR, Italy
Tel: +39 050 3153136
E-mail: anna.tonazzini![]() isti.cnr.it
isti.cnr.it