
 This issue in
This issue in 
Mining Genomic Data with Metaheuristic Techniques
by Carlos Cotta and Pablo Moscato
One of the major challenges in biomedical research is discovering the genetic basis of diseases such as Alzheimer's disease or schizophrenia. Research in the GISUM group at the University of Málaga is investigating the use of evolutionary metaheuristics for this purpose.
The pace at which research currently occurs is affecting most fields of science, not least the rapidly developing areas of molecular biology and genomics. The many initiatives in the life sciences that are planned or currently in execution are producing an unprecedented flood of data. As a result, many of the challenges in biology are increasingly becoming challenges in mathematics, and fundamentally in computing. The task of dealing with the large-scale combinatorial problems arising in bioinformatics is undoubtedly one of the greatest challenges facing computer science researchers, and new techniques and insights for algorithm design are required.
In molecular biology, the analysis of gene expression data represents one of these challenges. The difficulty of the problem lies in its computational complexity and the sheer amount of data that must be processed. For example, thanks to microarray technology we can monitor the activity of a whole genome in a single experiment. Huge amounts of data are becoming available thanks to this technique, providing access to a better picture of the simultaneous interactions of thousands of genes. The challenge now is to unravel the complex functional dependencies behind these data, and identify the linkage between genetic information and its phenotypical correlates.
From an algorithmic point of view, the data mining process can be shown in general to be hard according to the traditional P vs. NP scenario. For instance, assume that genomic data is available from both healthy individuals and patients affected by a certain pathology. Finding a minimal subset of genes such that the phenotypic status - healthy or ill - can be derived from their combined expression values is a problem for which no efficient (polynomial time) algorithm is known. This is just one example of the extremely hard tasks to be found in this domain.
In this situation, the classical approach is to define approximation algorithms. This is unpractical in many situations however, and has been superseded by two cutting-edge methodologies. In the first of these, parameterized complexity helps to identify tractable subclasses of these problems for a certain realistic range of some structural parameter of the problem; the resulting techniques are called fixed-parameter tractable (FPT) algorithms. In the second approach, modern heuristic techniques (metaheuristics) are employed to produce probably (though not yet provably) optimal solutions for these problems. The GISUM group of the University of Málaga (UMA) in Spain is working on this line of research in close cooperation with other centres worldwide, in particular the Newcastle Bioinformatics Initiative (NBI) in Australia. This cooperation has yielded numerous relevant results, and has been substantiated in an on-going project funded by the Australian Research Council on the area of genomic data mining with evolutionary algorithms (EAs).
We have been able to show that minimum feature-set selection problems are intrinsically hard beyond the NP sense. More precisely, our results indicate that no FPT algorithm is possible for this problem, thus emphasizing the prominent role that metaheuristics must play in this domain. Furthermore, given that the data are inherently noisy and prone to measurement errors, robust feature identification methods are essential. Our research has provided evidence that the joint use of evolutionary algorithms with a reduction approach inspired by kernelization rules often used in the design of FPT algorithms can provide good solutions for this problem, eg for discriminating between different types of lymphoma (see Figure 1).
|
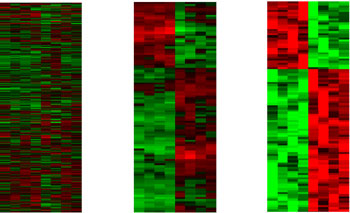
This methodology has also been deployed in conjunction with single-value decomposition and integer programming on microarray data from the brains of Alzheimer's patients and healthy patients used as a control. A clear pattern of differential gene expression is obtained, which can be regarded as a molecular signature of the disease. The results suggest that a unified approach may help to uncover complex genetic risk factors not currently discovered with a single method.
Our aims are now to formalize new problems in genomics as combinatorial, non-linear, mixed or multi-objective optimization problems and to study their computational complexity. Subsequently we plan to identify the best way of addressing and solving these problems using EAs and, where justified, hybridize the methods with exact algorithms and other types of metaheuristics. These techniques will be implemented in a unified software framework.
New collaborative initiatives are also underway. The most ambitious of these focuses on the use of bio-inspired techniques (subsuming EAs as well as artificial immune systems, or ant colony optimization) for mining genomic data, and involves institutions from Australia (NBI), France (Universities of Paris and Lille), the Netherlands (Free University Amsterdam), Spain (UMA) and the United Kingdom (University of Kent). We invite everyone to view our results and contact us with comments or suggestions for further collaboration.
Links:
http://www.lcc.uma.es/~ccottap
http://www.cs.newcastle.edu.au/~nbi/
Please contact:
Carlos Cotta
University of Málaga/SpaRCIM, Spain
Tel: +34 952 137158
E-mail: ccottap![]() lcc.uma.es
lcc.uma.es