
 This issue in
This issue in 
In Silico Virtual Experiments
by Wanda Andreoni
Using massively parallel computers and sophisticated mathematical modeling, scientists at IBM Zurich Research Lab have been able to shed new light on the binding of progesterone with its human receptor. Unraveling how progesterone binds to its human receptor is a first step in understanding that could lead to the development of more effective medication.
Progesterone, 'the hormone of pregnancy', is often the starting point for the design of so-called fertility drugs. For progesterone to have an effect it needs to get into cells. It can either diffuse through the cell membrane or bind to a protein in the cell membrane called a receptor. Controlling, activating, or blocking the action of this hormone amounts to modifying the binding with its receptor. So far, no methodology has been able to show unambigiously how this binding takes place, constituting a real puzzle.
|
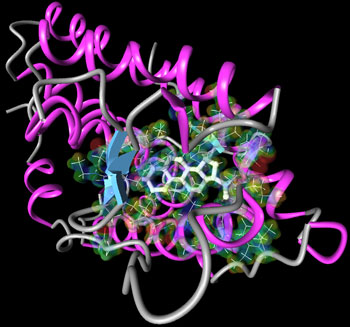
To study intermolecular interactions, the traditional tools are 'in vitro' (in a Petri glass) or 'in vivo' (in a living organism). Our in-silico method is effectively a simulation, based on classical molecular dynamics enhanced with quantum chemistry (quantum-refined-force-field molecular dynamics). The approach enables scientists to study the all-important role of water in the binding process, to analyze chemical and physical processes over time, and to show precisely how a given molecule (be it a hormone or a potential drug) and its biomolecular target (be it a protein or DNA) dynamically interact at the level of their component atoms.
The molecular structure of progesterone has two end groups that according to chemical intuition could contribute to the binding to its receptor. Contrary to indications from 'mutagenesis' analysis that confirmed this view, the X-ray structure of the complex has led to the belief that only one end was participating in the binding. This could be neither confirmed or refuted on the basis of traditional modeling.
Our in silico simulations have solved the puzzle and revealed that indeed in a typical aqueous solution both ends of progesterone play a crucial role in the binding in agreement with the results of mutagenesis analysis, and also revealed which parts of the protein (amino-acids) act as primary binding partners. They also showed the highly dynamic nature of the binding process, which is mediated by water molecules present in the pocket. The surprising results obtained from the X-ray structure could thus be ascribed to the sporadic presence of water in the crystal structure and also to the inability to reflect the flexibility of the chemical groups involved. Traditional modeling on the other hand lacked the accuracy that was necessary to represent the interatomic interactions of the real system.
The case of the progesterone-receptor complex is certainly not unique. Virtual experiments based on accurate simulations at the molecular level will increasingly be required to gain insight into the nature and functioning of complex systems like those we find in biology, and may indeed become of critical importance, given the unique information they can provide. As such they have also the potential to enhance rational drug design and thus to become a unique auxiliary tool in the development of pharmaceuticals, which could help reduce the time and expense associated with bringing new drugs to market.
It was fascinating to gain insight into the action of the hormone that determines human birth. Although still in its infancy, we believe that this synergetic approach of high performance computing and accurate methodologies for molecular simulations will lead the path into the unraveling of other mysteries of our life. Our research continues.
Link:
http://www.zurich.ibm.com/deepcomputing/scientific/projects_biochem.html
Please contact:
Wanda Andreoni,
IBM Zurich Research Lab/SARIT
E-mail: and![]() zurich.ibm.com
zurich.ibm.com