|
|
|
|||||||||
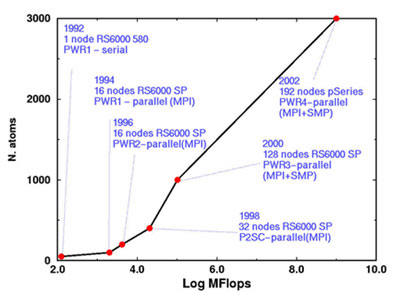
Towards the Design of Functional Materials and Drugs ‘from Scratch’by Wanda Andreoni and Alessandro Curioni High Performance Computing is a promising future technology for the design of novel materials including pharmaceuticals. Sustained innovation in computational methodologies is a key requisite for its success. Work in this direction is actively performed at the IBM Zurich Research Laboratory in Rüschlikon. Our daily life is dominated by innovations in materials and in drugs. One need only think of how the replacement of steel with aluminum alloys has made cars and airplanes lighter and faster, how our eyeglasses have become lighter and thinner thanks to the introduction of titanium for the lenses and titanium-rich metal alloys for the frames, how much brighter and finer the screens for our laptop displays are getting day after day as well as how much progress medicine has made over the years, thanks to the continuous development of new drugs. Over time the demands for new materials and drugs have changed, becoming much more specific and detailed. Replacing one material in a multicomponent device implies the optimization of many requirements under complex boundary conditions; for example, a novel material that may one day replace silicon dioxide as gate dielectric in CMOS technology not only has to provide higher permittivity but also has to preserve sufficiently high electron mobility and be integrable in current processing technology. Drug design has always been known to be extremely complex, but lately it has become clear that real progress will only be possible through personalized medicine. Given that the need is increasingly more sophisticated, one must rely on increasingly powerful and sophisticated design methodologies and on their sustained innovation. Computers nowadays have the power to store several terabytes of data and to process them at the speed of 10-20 teraflops. As this power becomes more widely available, the door opens for the creation of powerful simulation techniques. Their development is being pursued by several groups in well-known research and industrial institutions worldwide. The ultimate goal of this global effort is that of creating a virtual laboratory in which new compounds are synthesized, screened and optimized through the direct observation of their evolution in time in response to the change of physical conditions (eg, temperature and pressure) and/or to the action of chemical agents. At the IBM Zurich Research Laboratory, the Computational Biochemistry and Materials Science Group is active in the development of accurate and efficient methodologies for the simulation of diverse materials, ranging from silicon to enzymes, at the atomistic level. The specific purpose of this work is that of obtaining the above-mentioned lead optimization ‘from scratch’, namely, guided only by the knowledge of the fundamental parameters of the atomic components, and from the direct computation of the intra- and intermolecular interactions based on the laws of quantum mechanics. A fully quantum-mechanical approach to a given compound has long been hindered by the limited size (number of atoms) of the model. The required analysis is becoming feasible through modern computing power; the upper limit to the size of this model is bound to increase over the years with the increase of the power of the hardware and the simultaneous progress in the algorithms. As an example, Figure 1 shows how the size of the systems one can simulate from first-principles has changed in the past decade with the increase in computer power. The leap forward in 1994 was due to the advent of parallel computers and the use of a highly parallelized code. The parallelization of such a code is a non-trivial task since it is composed of several parts that scale non-homogeneously with the system size and the dominant parts (both in terms of memory use and computational time) which are explicitly dependent on system size, ie, elements, that dominate the computation at small/intermediate sizes, become irrelevant at larger sizes. In particular, there are two different classes of algorithms in the code: 1) Fast Fourier Transform related algorithms (which scale as V2logV, where V is the ‘volume’ of the system) and 2) Linear algebra related algorithms (such as orthogonalization, that scale as V3). At small/intermediate system sizes, type 1 algorithms are dominant and these were parallelized in our laboratory in 1994 using an MPI (Message Passing Interface) implementation on distributed memory computers. Type 2 algorithms, however, only become important for molecular systems containing more than 1000 atoms and were parallelized with OpenMP (Open Message Passing) on mixed shared/distributed memory computers in 2000. These two steps were instrumental in extending the application realm of our simulation techniques. For an extended description of the method see W.Andreoni and A.Curioni, ‘New Advances in Chemistry and Material Science with CPMD and Parallel Computing’, Parallel Computing 26 (2000) 819.
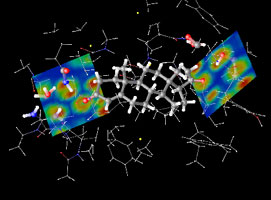
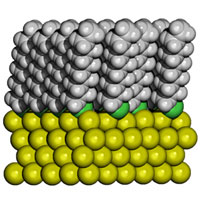
Currently our simulations of large systems (thousands of atoms) rely mostly on a kind of hybrid modelling in which that part of the compound where ‘the action takes place’ is treated at the quantum level and the rest (‘the environment’) at the classical level, namely with interactions modeled using semi-empirical parameters. The former can go up to sizes of ~1000 atoms, but calculations would not achieve the required degree of accuracy if the effects of the interactions with its environment were neglected. Examples of biochemical processes we have studied in this way are enzymatic reactions in which the chemical event activated by the presence of the enzyme is relatively confined, and the binding of ligands to protein (receptors) in aqueous solution (Figure 2), which is related to the design of optimal drugs. Examples of advanced materials for technological use are the organic components of light-emitting devices (LED), namely amorphous molecular compounds interacting with the electrodes, and functionalized nanostructures of self-assembled monolayers of organic chains on metal substrates (Figure 3) that are used in diverse technological applications such as the fabrication of sensors and transducers and as patternable materials. The success of High Perfomance Computing-driven design clearly depends on the steady increase of computational capabilities. One usually refers to Moore’s law, which predicts that transistor density doubles every 18 months. It is no secret, however, that IT industries are currently struggling to maintain this pattern because of the intrinsic physical limits of the technology. As history teaches us, crises are often overcome by the advent of new concepts. A most promising one is that of GRID computing. Link: Please contact: |
||||||||||