

by Alexander Zien, Robert Küffner, Theo Mevissen, Ralf Zimmer and Thomas Lengauer
As ever more knowledge is accumulated on various aspects of the molecular machinery underlying the biological processes within organisms, including the human, the question how to exploit this knowledge to combat diseases becomes increasingly urgent. At GMD scientists work to utilize protein structures, expression profiles as well as metabolic and regulatory networks in the search for target proteins for pharmaceutical applications.
Huge amounts of heterogeneous data are pouring out of the biological labs into molecular biology databases. Most popular are the sequencing projects that decipher complete genomes and uncover their protein complements. Many more projects are under way, aiming eg at resolving the yet unknown protein folds or at collecting human single nucleotide polymorphisms. In a less coordinated way, many labs measure gene expression levels inside cells in numerous cell states. Last but not least there is an enormous amount of unstructured information hidden in the plethora of scientific publications, as is documented in PubMed, for instance. Each of these sources of data provides valuable clues to researchers that are interested in molecular biological problems. The project TargId at GMD SCAI focuses on methods to address the arguably most urgent problem: the elucidation of the origins and mechanisms of human diseases, culminating in the identification of potential drug target proteins.
TargId responds to the need for bioinformatics support for this task. The goal of the project is to develop methods that extract useful knowledge from the raw data and help to focus on the relevant items of data. The most sophisticated aspect is the generation of new insights through the combination of information from different sources. Currently, our TargId methodology builds on three main pillars: protein structure prediction, expression data analysis and metabolic/regulatory pathway modeling.
Protein Structure Prediction
Knowledge on the three-dimensional structure (fold) of a protein provides clues on its function and aids in the search for inhibitors and other drugs. Threading is an approach to structure prediction which essentially assesses the compatibility of the given protein sequence to a known fold by aligning the sequence onto the known protein structure. Thus, threading utilizes the available knowledge directly in the form of the structures that are already experimentally resolved. Another advantage of threading in comparison to ab initio methods is the low demand of computing time. This is especially true for 123D, a threader developed at GMD that models pairwise amino acid contacts in a way that allows alignment computation by fast dynamic programming. The objective function for the optimization includes potentials accounting for chemical environments within the protein structure, and membership in secondary structure elements as well as amino acid substitution scores and gap penalties, all carefully balanced by a systematic procedure. A second threading program programmed at GMD, called RDP, utilizes more computation time than 123D in order to optimize full pair interaction contact potentials to yield refined alignments.
Expression Data Analysis
Data on expressed genes comes in several different flavors. The historically first method is the generation of ESTs (expressed sequence tags), i.e. low-quality sequence segments of mRNA, either proportional to its cellular abundance or enriched for rare messages. While ESTs are superseded by more modern methods for the purpose of expression level measurements, they are still valuable for finding new genes and resolving gene structures in the genomic DNA. Thus, we have implemented a variant of 123D that threads ESTs directly onto protein structures, thereby translating nucleotide codons into amino acids on the fly. This program, called EST123D, is useful for proteins that are not yet characterized other than by ESTs.
Nowadays, gene expression levels are most frequently obtained by DNA chips, micro-arrays or SAGE (serial analysis of gene expression). These technologies are designed for high throughput and are already capable of monitoring the complete gene inventory of small organisms (or a large part of all human genes). We apply statistics and machine learning techniques in order to normalize the resulting raw measurement data, to identify differentially regulated genes and to clusters of cell states. Subsequently, we apply statistics and machine learning techniques in order to identify differentially regulated genes, clusters of cell states etc.
Pathway Modeling
While a large part of the current effort is focused on inferring high-level structures from gene expression data, much is already known on the underlying genetic networks. Several databases that are available on the internet document metabolic relations in machine-readable form. The situation is worse for regulatory pathways; most of this knowledge is still hidden in the literature. Consequently, we have implemented methods that extract additional protein relations from article abstracts and model them as Petri nets. The resulting graphs can be restricted to species, tissues, diseases or other areas of interest. Tools are under development for viewing and editing using standard graph and Petri net packages. The generated networks can provide overviews that cross the boundaries of competence fields of human experts.
Further means are necessary to allow for more detailed analyses. We can automatically extract pathways from networks that are far too large and complicated to lend themselves to easy interpretation. Pathways are biologically meaningful subgraphs, eg signaling cascades or metabolic pathways that account for supply and consumption of any intermediate metabolites. Another method conceived in TargId, called DMD (differential metabolic display), allows for comparing different systems (organisms, tissues, etc.) on the level of complete pathways rather than mere interactions.
Bringing it All Together ...
Each of the methods described above can provide valuable clues pointing to target proteins. But the crux lies in their clever combination, interconnecting data from different sources. In recent work, we have shown that in real life situations clustering alone may not be able to reconstruct pathways from gene expression data. Instead of searching for meaning in clusters, we invented an approach that proceeds inversely: First, a set of pathways is extracted from a protein/gene network, using the methods described above. Then, these pathways are scored with respect to gene expression data. The restriction to pathways prevents us from considering unreasonable groupings of proteins, while it still allows for incorporating and testing hypotheses. Eg, pathways can be constructed from interactions that are observed in different tissues or species. The expression data provide an orthogonal view on these interactions and can thus be used to validate the hypotheses.
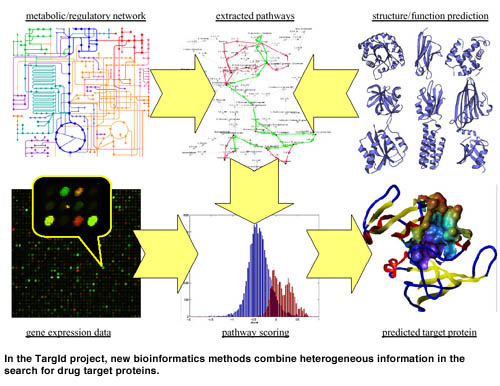
Structure prediction can aid in this process at several stages. First, uncharacterized proteins can tentatively be embedded into known networks based on predicted structure and function. Second, structural information can be integrated into the pathway scoring function. Finally, when a target protein is identified, its structure will be of utmost interest for further investigations.
It can be imagined that target finding can gain from broadening the basis for the search to also include, eg, phylogenetic profiles, post-translational modifications, genome organization or polymorphisms. As these fields are still young and in need of further progress, it is clear that holistic target finding is only in its infancy.
Links:
http://cartan.gmd.de/TargId/
Please contact:
Alexander Zien or Ralf Zimmer - GMD
Tel: + 49 2241 14-2563 or -2818
E-mail: Alexander.Zien@gmd.de or Ralf.Zimmer@gmd.de