

by Marie-France Sagot
It is almost a ‘cliché’ to say that the sequencing at an everincreasing speed of whole genomes, including that of man, is generating a huge amount of information for which the use of computers becomes essential. This is true but is just a small part of the truth if one means by that the computer’s capacity to physically store and quickly sift through big quantities of data. Computer science may strongly influence at least some areas of molecular biology in other, much deeper ways.
A lot of the problems such areas have to address, in the new post-sequencing era but not only, requires exploring the inner structure of objects, trying to discover regularities that may lead to rules, building general models which attempt to weave and exploit relations among objects and, finally, obtaining the means to represent, test, question and revise the elaborated models and theories. These are all activities that are at the essence of most currently elaborated computer algorithms for analysing, often in quite subtle ways, biological data.
Among the algorithmical approaches possible, some try to exhaustively explore all the ways a molecular process could happen given certain (partial) beliefs we have about it. This leads to hard combinatorial problems for which efficient algorithms are required. Such algorithms must make use of complex data representations and techniques.
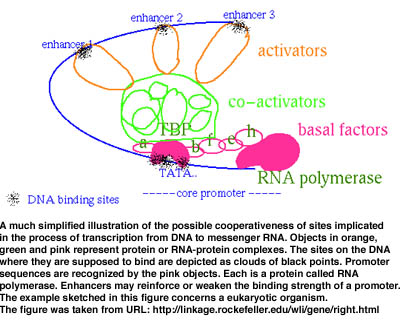
For instance, quite a few processes imply the recognition by the cell machinery of a number of sites along a DNA sequence. The sites represent fragments of the DNA which other macromolecular complexes must be able to recognize and bind to in order for a given biological event to happen. Such an event could be the transcription of DNA into a so-called messenger RNA which will then be partially translated into a protein. Because their function is important, sites tend to be conserved (though the conservation is not strict) while the surrounding DNA suffers random changes. Other possible sequential and/or spatial characteristics of sites having an equivalent function in the DNA of an organism may exist but are often unknown. Hypotheses must then be elaborated on how recognition works. For example, one probable scenario is that sites are cooperative as suggested in Figure 1. Each one is not recognized alone but in conjunction with others at close intervals in space and time. A model based upon such a hypothesis would try to infer not one site (corresponding to inferring the presence of a conserved word at a number of places along a string representing a DNA sequence), but various sites simultaneously, each one located at an often precise (but unknown) distance from its immediate neighbours (a problem one may see as equivalent to identifying a maximal common sequence of conserved and spatially constrained motifs). The model may be further sophisticated as the analysis progresses. What is important to observe is that the revision of a model may be effected only if the algorithm based upon it is exhaustive.
Exact algorithms may therefore be powerful exploratory tools even though they greatly simplify biological processes (all approaches including experimental ones, do). In particular, exact algorithms allow to ‘say something’, derive some conclusions even from what is not identified because we know that, modulo some parameters they may take as input, they exhaustively enumerate all objects they were built to find. Whether they indeed find something (for instance, a site initially thought to exist with a certain characteristic) or not, we may thus, by understanding their inner workings, emit biological hypotheses on ‘how things function’, or do not function. This is possible also with non exact approaches such as some of the statistical ones, but is often more difficult to realize.
A first algorithm using a model which allows to simultaneously infer cooperative sites in a DNA sequence has been elaborated (in C) by the algorithmics group at the Pasteur Institute (composed of one researcher and PhD students from the Gaspard Monge Institute). In collaboration with biology researchers from the Biology and Physico-Chemistry and the Pasteur Institutes in Paris, this algorithm has started been applied to the analysis of the sequenced genomes of three bacteria: Escherichia coli, Bacillus subtilis and Helicobacter pylori. The objective was to identify the main sequence elements involved in the process of transcription of DNA into messenger RNA (what is called the promoter sequences). For various reasons, it was suspected that such elements would be different in Helicobacter pylori as compared to the other two bacteria. A blind, exhaustive inference of single conserved words in the non coding regions of the genome of Helicobacter pylori permitted to identify some elements but not to put together with enough certainty those implicated in the transcriptional process. The development of more complex models was called upon and, indeed, allowed to reveal that the sequence elements are not the same in Helicobacter pylori. It has also enabled to suggest a consensus for what the main so-called promoter elements should be in the bacterium. It was later verified that the consensus proposed strongly resembles one experimentally determined for a bacterium which is closely related in terms of evolution to Helicobacter pylori.
This particular algorithm is been continously ameliorated, expanded and extended to take into account the increasingly more sophisticated models which further analyses of other genomes and processes reveal are necessary to get closer to the way recognition happens in diverse situations. Other combinatorial algorithms, addressing a variety of subjects – molecular structure matching, comparison and inference; regularities detection; gene finding; identification of recombination points; genome comparison based upon rearrangement distances – are currently been studied within the Pasteur Institute algorithmics group, often in collaboration with outside PhD students and researchers.
It is worth observing that many computer scientists come to biology seduced by the huge range of nice problems that this offers to them without necessarily wanting to get implicated into the intricacies of biology itself. Although the two, computer science and biology, remain quite distinct fields of research, it appears difficult to gain maturity and any real insight into the domain without getting further involved into biology. This is particularly, but not exclusively, true if one adopts a combinatorial approach as the operations of building models that try to adhere to the way biological processes happen, and algorithms for exploring and testing such models, become intimately linked in a quasi-symbiotic relationship. It would, anyway, be a pity not to get so involved as the problems become much more interesting, even from a purely computational and combinatorial point of view, when they are not abstracted from what gave origin to them: a biological question. The case of identifying sites is just one example among many.
Links:
Papers by the group on this and other combinatorial problems: http://www-igm.univ-mlv.fr/~sagot
Series of seminars on this and other topics: http://www.pasteur.fr/infosci/conf/AlgoBio
Please contact:
Marie-France Sagot - Institut Pasteur
Tel: + 33 1 40 61 34 61
E-mail: sagot@pasteur.fr