

by Matthias Rarey and Thomas Lengauer
The rapid progress in sequencing the human genome opens the possibility for the near future to understand many diseases better on molecular level and to obtain so-called target proteins for pharmaceutical research. If such a target protein is identified, the search for those molecules begins which influence the protein’s activity specifically and which are therefore considered to be potential drugs against the disease. At GMD, approaches to the computer-based search for new drugs are being developed (virtual screening) which have already been used by industry in parts.
Searching for New Lead Structures
The development process of a new medicine can be divided into three phases. In the first phase, the search for target proteins, the disease must be understood on molecular-biological level as far as to know individual proteins and their importance to the symptoms. Proteins are the essential functional units in our organism and can perform various tasks ranging from the chemical transformation of materials up to the transportation of information. The function is always linked with the specific binding of other molecules. As early as 100 years ago, Emil Fischer recognised the lock-and-key principle: Molecules that bind to each other are complementary to each other both spatially and chemically, just as only a specific key fits a given lock (see Figure 1). If a relationship between the suppression (or reinforcement) of a protein function and the symptoms is recognised, the protein is declared to be a target protein. In the second phase, the actual drug is developed. The aim is to detect a molecule that binds to the target protein, on the one hand, thus hindering its function and that, on the other, has got further properties that are demanded for drugs, for example, that it is well tolerated and accumulates in high concentration at the place of action. The first step is the search for a lead structure - a molecule that binds well to the target protein and serves as a first proposal for the drug. Ideally, the lead structure binds very well to the target protein and can be modified such that the resulting molecule is suitable as a drug. In the third phase, the drug is transformed into a medicine and is tested in several steps to see if it is well tolerated and efficient. The present paper is to discuss the first step, ie the computer-based methods of searching for new lead structures.
New Approaches to Screening Molecule Databases
The methods of searching for drug molecules can be classified according to two criteria: the existence of a three-dimensional structural model of the target protein and the size of the data set to be searched. If a structural model of the protein is available, it can be used directly to search for suitable drugs (structure-based virtual screening); ie we search for a key fitting a given lock. If a structural model is missing, the similarity to molecules that bind to the target protein is used as a measure for the suitability as a drug (similarity-based virtual screening). Here we use a given key to search for fitting keys without knowing the lock. In the end, the size of the data set to be searched decides on the amount of time to be put into the analysis of an individual molecule. The size ranges from a few hundred already preselected molecules via large databases of several millions of molecules to virtual combinatorial molecule libraries theoretically allowing to synthese of up to billions of molecules from some hundred molecule building blocks.
The key problem in structure-based virtual screening is the prediction of the relative orientation of the target protein and a potential drug molecule, the so-called docking problem. For solving this problem we have developed the software tool FlexX [1] in co-operation with Merck KGaA, Darmstadt, and BASF AG, Ludwigshafen. On the one hand, the difficulty of the docking problem arises from the estimation of the free energy of a molecular complex in aqueous solution and, on the other, from the flexibility of the molecules involved. While a sufficient description of the flexibility of the protein presumably will not be possible even in the near future, the more important flexibility of the ligand is considered during a FlexX prediction. In a set of benchmarks tests, FlexX is able to predict about 70 percent of the protein-ligand complexes sufficiently similar to the experimental structure. With about 90 seconds computing time per prediction, the software belongs to the fastest docking tools currently available. FlexX has been marketed since 1998 and is currently being used by about 100 pharmaceutical companies, universities and research institutes.
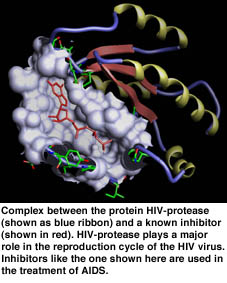
If the three-dimensional structure of the target protein is not available, similarity-based virtual screening methods are applied to molecules with known binding properties, called the reference molecule. The main problem here is the structural alignment problem which is closely related to the docking problem described above. Here, we have to superimpose a potential drug molecule with the reference molecule so that a maximum of functional groups are oriented such that they can form the same interactions with the protein. Along the lines of FlexX, we have developed the software tool FlexS for the prediction of structural alignments with approximately the same performance with respect to computing time and prediction quality.
If very large data sets are to be searched for similar molecules, the speed of the alignment-based screening does not suffice yet. The aim is to have comparison operations whose computation takes by far less than one second. Today linear descriptors (bit strings or integral vectors) are usually applied to solve this problem. They store the occurrence or absence of characteristic properties of the molecules such as specific chemical fragments or short paths in the molecule. Once such a descriptor has been determined, the linear structure enables a very fast comparison. A considerable disadvantage is, however, that the overall structure of the molecule is represented only inadequately and an exact match between the fragments is frequently required for the recognition of similarities. As an alternative, we have developed a new descriptor, the feature tree [4], in co-operation with SmithKline Beecham Pharmaceuticals, King of Prussia (USA) . Unlike the representation using linear descriptors, in this approach a molecule is represented by a tree structure representing the major building blocks of the molecules. If two molecules are to be compared with each other, the task is to find first an assignment of the building blocks of the molecules which might be able to occupy the same regions of the active site upon binding. With the aid of a time-efficient implementation, average comparison times of less than a tenth second can be achieved. This allows 400.000 molecule comparisons to be carried out overnight within a single computation on a single processor. Applying the new descriptor to benchmark data sets, we could show that, in many cases, an increase of the rate of active molecules in a selected subset of the data set is achieved if compared with the standard method.
Designing New Medicines with the Computer?
Unlike many design problems from the field of engineering, for example, the design of complex plants, machines or microchips, the underlying models are still very inaccurate for solving problems in the biochemical environment. Important quantities such as the binding energy of protein-ligand complexes can be predicted only with high error rates. In addition, not only on the interaction of the drug with the target protein is of importance to the development of medicines. The influence of the drug on the whole organism rather is to be examined. Even in the near future, it will not be possible to answer many questions arising in this context accurately by means of the computer due to their complexity: Is the drug absorbed in the organism? Does it produce the desired effect? Which side effects are experienced or is it possibly even toxic? Therefore a medicine originating directly from the computer will not be available neither in the near nor in the distant future. Nevertheless, the importance of the computer increases in drug research. The reason is the very great number of potential molecules which come into consideration as a drug for a specific application. The computer allows a reasonable pre-selection and molecule libraries can be optimised for specific characteristics before synthesising. On the basis of experiments, the computer is able to generate hypotheses which enable in turn better planning of further experiments. In these domains, the computer has already proven to be a tool without which pharmaceutical research cannot be imagined anymore.
Links:
http://www.gmd.de/SCAI/
Please contact:
Matthias Rarey - GMD
Tel: +49 2241 14 2476
E-mail: matthias.rarey@gmd.de