

by Anton Enright, Sophia Tsoka and Christos Ouzounis
The current goal of bioinformatics is to take the raw genetic information produced by sequencing projects and make sense of it. The entire genome sequence should reflect the inheritable properties of a given species. At the Computational Genomics Group of the European Bioinformatics Institute (an EMBL outstation) in Cambridge, work is underway to tackle this vast flood of data using both existing and novel technologies for biological discovery.
The recent sequencing of the complete genomes of many species (including a ‘draft’ human genome) has emphasised the importance of bioinformatics research. Once the DNA sequence of an organism is known, proteins encoded by this sequence are predicted. While some of these proteins are highly similar to well-studied proteins whose functions are known, many will only have similarity to another poorly annotated protein from another genome or worse still, no similarity at all. A major goal of computational genomics is to accurately predict the function of all proteins encoded by a genome, and if possible determine how each of these proteins interacts with other proteins in that organism. Using a combination of sequence analysis, novel algorithm development and data-mining techniques the Compuational Genomics Group (CGG) is targeting research on the following fields.
Automatic Genome Annotation
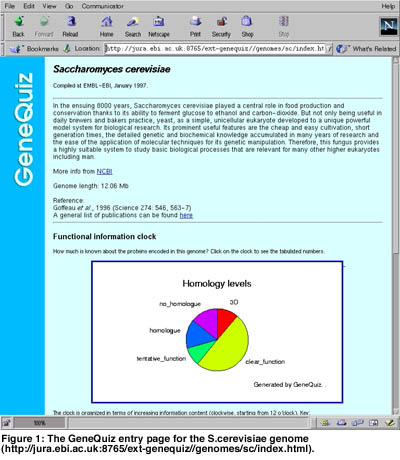
Accurately annotating the proteins encoded by complete genomes in a comprehensive and reproducible manner is important. Large scale sequence analysis necessitates the use of rapid computational methods for functional characterisation of molecular components. GeneQuiz is an integrated system for the automated analysis of complete genomes that is used to derive protein function for each gene from raw sequence information in a manner comparable to a human expert. It employs a variety of similarity search and analysis methods that entail the use of up-to-date protein and DNA databases and creates a compact summary of findings that can be accessed through a Web-based browser. The system applies an ‘expert system’ module to assess the quality of the results and assign function to each gene.
Assigning Proteins into Families
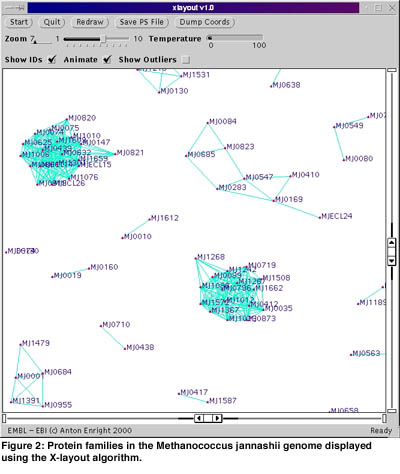
Clustering protein sequences by similarity into families is another important aspect of bioinformatics research. Many available clustering techniques fail to accurately cluster proteins with multiple domains into families. Multi-domain proteins generally perform at least two functions that are not necessarily related, and so ideally should belong in multiple families. To this end we have developed a novel algorithm called GeneRAGE. The GeneRAGE algorithm employs a fast sequence similarity search algorithm such as BLAST and represents similarity information between proteins as a binary matrix. This matrix is then processed and passed through successive rounds of the Smith-Waterman dynamic programming algorithm, to detect inconsistencies which represent false-positive or false-negative similarity assignments. The resulting clusters represent protein families accurately and also contain information regarding the domain structure of multi-domain proteins. A visualization program called xlayout based on the Fruchterman-Rheingold graph-layout optimisation algorithm has also been developed for displaying these complex similarity relationships.
Prediction of Protein Interaction
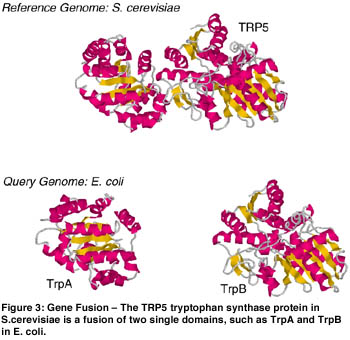
Another novel algorithm developed in the CGG group is the DifFuse algorithm. This algorithm is based on the hypothesis that there is a selective advantage for proteins performing related functions to fuse together during the course of evolution (eg different steps in the same metabolic pathway). The DifFuse algorithm can detect a fused protein in one genome based its similarity to complementary pair of unfused proteins in another genome. The detection of these fused proteins allows one to predict either functional association or direct physical interaction of the un-fused proteins. This algorithm is related to GeneRAGE in the sense that the fusion detection process is similar to the multi-domain detection step described above. This algorithm can be applied to many genomes for large-scale detection of protein interactions.
Knowledge-Base Development
Databases in molecular biology and bioinformatics are generally poorly structured, many existing as flat text files. In order to get the most out of complex biological databases these data need to be represented in a format suitable for complex information extraction through a simple querying system and also ensure data integrity. An ontology is an exact specification of a data model that can be used to generate such a ‘knowledge’ base. We have developed an ontology for representation of genomic data which is used to built a database called GenePOOL incorporating these concepts. This system stores computationally-derived information such as functional classifications, protein families and reaction information. Database analysis is performed through flexible and complex queries using LISP that are simply not possible through any other public molecular biology database. Similarly, we have also developed a standard for genome annotation called GATOS (Genome AnoTattiOn System) which is used as a data exchange format. Work is under development to incorporate an XML-based standard called XOL (XML Ontology Language).
Text-Analysis and Data-Mining
There is already a vast amount of data available in the abstracts of published biological text. The MEDLINE database contains abstracts for over 9 million biological papers published worldwide since 1966. However, these data are not represented in a format suitable for large-scale information extraction. We have developed an algorithm called TextQuest which can perform document clustering of MEDLINE abstracts. TextQuest uses an approach that restructures these biological abstracts and obtains the optimal number of terms that can associate large numbers of abstracts into meaningful groups. Using a term-weighting system based on the TF.IDF family of metrics and term frequency data from the British National Corpus, we select words that are biologically significant from abstracts and add them to a so-called go-list. Abstracts are clustered using an unsupervised machine learning approach, according to their sharing of words contained in the go-list. The xlayout algorithm (see above) is then used to display the clustering results. The resulting document clusters accurately represent sets of abstracts referring to the same biological process or pathway. TextQuest has been applied to the development of the dorsal-ventral axis in the fruit-fly Drosophila melanogaster and has produced meaningful clusters relating to different aspects of this developmental process.
Links:
http://www.ebi.ac.uk/research/cgg/
Please Contact:
Christos A. Ouzounis – European Molecular
Biology Laboratory, European Bioinformatics Institute
Tel: +44 1223 49 46 53
E-mail: ouzounis@ebi.ac.uk