PROTAL - Protein Structure Prediction and Accurate Alignment of Protein Sequences and Structures
by Heinz Mevissen, Joachim Selbig, Ralf Thiele, Ralf Zimmer and Thomas Lengauer
The PROTAL project focuses on the development of computer-aided, theoretical prediction methods for structure and function of given protein sequences. The three-dimensional structure of proteins determines their biochemical function in organisms in a complex process network of interactions and regulations. Theoretical prediction methods try to shortcut labour-intensive, time-consuming and expensive experimental structure determination in order to reduce the increasing gap between the large number of of sequenced proteins produced within genome and large scale sequencing projects and known protein structures. Especially, predictions and modeling of binding and active sites of particular proteins are of interest for biotechnology and pharmaceutical industry.
The PROTAL project aims at the development of scoring schemes and methods to support the construction of structural models for protein sequences based on the homology modeling paradigm, ie inferring partial structures from similarities with experimentally determined protein structures. The scoring schemes and algorithmic methods are integrated in a software package ToPLign (TOolbox for Protein aLIGNment).
The PROTAL project develops new hypotheses and models for factors determining protein structure, which are essential as scoring schemes for prediction methods. Especially new empirical potentials for amino acid interactions are derived from databases of experimentally determined structures. In order to appropriately model such interactions we developed a new contact definition based on Voronoi tesselations of protein structures. The potentials are used for the identification of appropriate targets as well as the computation of structurally compatible alignments.
Protein fold recognition allows the rapid identification of evolutionary, structurally, or functionally related proteins of known structure, so called targets. Accurate homology modeling of such proteins requires high quality alignments of the sequences in question to identified target structures. Refined alignments specify the coordinates of identically matched amino acids and determine amino acid side chains to be placed and backbone loop regions to be modeled.
A significant sequence similarity between sequences of unknown structure and known protein families is demonstrated via extended and new methods for the visualization and reliability of alignments, for tree and clustering multiple profile alignments, and for the computation of all compatible phylogenetic trees together with their associated multiple alignments. New threading approaches are developed in order to show sequence-structure compatibility: The 123D threading method computes optimal alignments with respect to so-called contact capacity potentials (CCP). These potentials represent a detailed measure for hydrophobicity and contact environments. The RDP (recursive dynamic programming) threading algorithm is used to refine computed alignments and structural models. The method concentrates on the most important regions of the protein and optimizes the mapping recursively. The procedure optimally extends subalignments by dynamic programming with respect to a full pair interaction potential of already mapped parts.
For all alignment and threading methods the inherent inaccuracy of biological data has to be taken into account in the methods and their statistical validation. In order to analyse the dependency of alignments on the parameters of the scoring scheme we use a new parametric optimization method, which is able to compute, for a given algorithm and type of scoring function, all different optimal alignments over the whole range of parameter settings. This allows for the systematic evaluation of competing methods and for insights into the reliabilty of computed alignments.
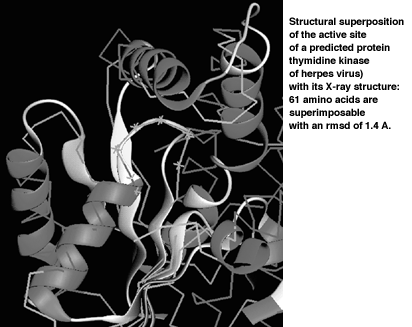
The Figure shows our successful ToPLign prediction of the structure for the active site of the thymidine kinase of the herpes virus, which is important for the development of inhibitors. The sequence was kindly provided by BASF AG. The developed methods have also been successfully applied in an international competition to predict the structure of proteins, which are close to being solved experimentally.
A prototype of the alignment tool ToPLign, including the fast threading method 123D and the RDP method, has been distributed since mid 1994 and been updated since then. The software is also available via the World Wide Web and mirrored at the National Cancer Institute (NCI/NIH) in Frederick, MD, USA. It is in use in various university institutes and some US biotechnology companies.
Please contact:
Heinz Mevissen - GMD
Tel: +49 2241 14 2784
E-mail: heinz-theodor.mevissen@gmd.deJoachim Selbig - GMD
Tel: +49 2241 14 2792
E-mail: joachim.selbig@gmd.deRalf Thiele - GMD
Tel: +49 2241 14 2302
E-mail: ralf.thiele@gmd.deRalf Zimmer - GMD
Tel: +49 2241 14 2818
E-mail: ralf.zimmer@gmd.deThomas Lengauer - GMD
Tel: +49 2241 14 2777
E-mail: thomas.lengauer@gmd.de