GMD Successful at a World-Wide Protein Structure Prediction Experiment
by Bernd Kramer and Thomas Lengauer
Seventy-two research groups from all over the world participated in the Second Meeting on the Critical Assessment of Techniques for Protein Structure Prediction (CASP2) at Asilomar, California, 12-16 December 1996. This meeting was the culmination of a world-wide experiment to determine the effectiveness of current structure prediction methods. The two projects PROTAL (Proteins: Sequence, Structure, and Evolution) and RELIWE (Calculation and Prediction of Receptor Ligand Interactions) from the Institute for Algorithms and Scientific Computing at GMD participated in this experiment and exhibited successful predictions.
The aim of the CASP2 experiment was to test existing structure prediction methods and software tools on so-called blind predictions. A blind prediction is a structure prediction, for which the actual structure is not known at the time of the prediction, but will become available soon thereafter for evaluation. The organizing team of CASP2 at Lawrence Livermore Laboratory, California, had collected 42 different prediction targets from protein crystallographers and Nuclear Magnetic Resonance (NMR) spectroscopists, who provided the sequences for prediction before the summer of 1996, and the resolved structures lateron, as they became available in late 1996. During the intervening months the predictor teams submitted structure models based on their theoretical methods. At the last deadline for submission of the predictions, more than nine hundred models had been sent to the organizers.
There were four disciplines within the contest:
Comparative Modeling
Here the protein sequence given displays a high degree of homology (above 40% sequence identity) to a protein of known structure. The goal is to generate a detailed atomic structure model of the protein.
Threading
Here the protein sequence given displays a low or marginal similarity (less than 40% and down to well below 20% sequence identity) to a structurally known protein. The goal is twofold. First, detect a good structural model - the so-called template - of the protein in question -the so-called target - among the proteins whose structure is known. This task is called fold recognition. The second goal is to faithfully map the target sequence onto the template structure. This task is called threading.
Ab Initio Prediction
Here the structure prediction of the sequence in question is not based on a homologue among the structurally known proteins, most often because no such homologue exists. In this case, one should find out whatever one can about the protein structure, eg, in terms of secondary structure, topology, or tertiary structure.
Docking
Here, the available information includes a structure of a protein and a structural formula of a ligand molecule that binds to the protein. The ligand can be another protein (in which case its structure is given, as well) or a small molecule. The goal is to predict the structure of the molecular complex consisting of the protein and the bound ligand.
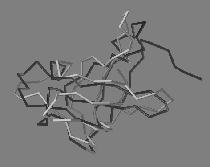
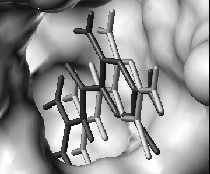
Predictions (light grey) of a protein structure (left) and a ligand position (right), experimental structures in dark grey.
GMD groups in competition
The two groups of GMD Institute for Algorithms and Scientific Computing entered this competition in the disciplines fold recognition (threading) and docking.
The PROTAL group (http://cartan.gmd.de/PROTAL/) has developed the program package ToPLign (http://cartan. gmd.de/ToPLign.html) which can be used to analyze, and align protein sequences and predict protein structures on the basis of their sequence. One of the tools developed within this suite is an improved follow-up of the predictor 123D which in a recent report in Nature Structural Biology has been mentioned as a prime resource for threading. Another tool called RDP, included in the ToPLign package can be used to refine these alignments in order to reach a model quality that is sufficient for docking studies. In the project RELIWE (http://www.gmd.de/SCAI/alg/reliwe/reliwe_home.html) the development of algorithms for docking of flexible ligand molecules at GMD has led to the software tool FlexX, the fastest docking tool published so far, and apparently the only tool currently available on the internet (http://cartan.gmd.de/FlexX.html).
FlexX supports ligand flexibility, handles steric as well as chemical aspects of docking and produces models whose quality is comparable to that of other tools that use much more runtime.
With the help of these prediction tools the two GMD groups have submitted models for most of the targets within the disciplines (2) and (4) of CASP-2. At the meeting the methods and the results were presented, and a team of independent scientists made an assessment of the quality of the predictions. PROTAL and RELIWE turned out to be well placed among the leading groups of both prediction areas. In particular, the reliability of the predictions was high, the number of false positives in fold recognition was low, and the docking tool FlexX had the smallest runtimes among all docking tools.
Please contact:
Bernd Kramer - GMD
Tel: +49 2241 14 2276
E-mail: bernd.kramer@gmd.deThomas Lengauer - GMD
Tel: +49 2241 14 2777
E-mail: thomas.lengauer@gmd.de